
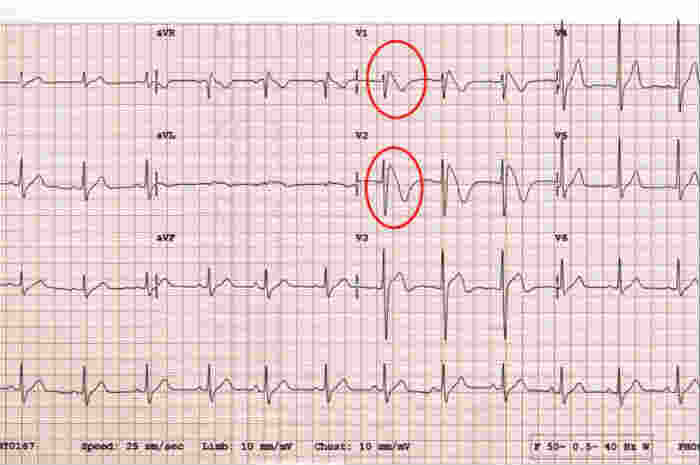
La Sindrome di Brugada è un'aritmia cardiaca causata da una mutazione genetica (SCN5A, ma non esclusivamente) presente sul cromosoma 3. È una malattia aritmogena a trasmissione autosomica dominante caratterizzata da un tipico ECG (segno di Brugada con sopraslivellamento del tratto ST in V1 e V3 e blocco di branca destra) e da sincope e/o morte improvvisa.
Paradosso Brugada: Per scoprirla ci vuole un arresto cardiaco
Brugada
Suscitano sempre grande interesse e curiosità le nuove scoperte, ma anche un po' di timore per i vari aspetti ancora da definire e approfondire.
Una di queste ha attirato particolarmente la mia attenzione sia per la mancanza di criteri diagnostici univoci sia per le conoscenze ancora incomplete sulla malattia.
Infine anche per la gravità degli effetti e per l'assenza di un approccio farmacologico efficace che possa ridurre significativamente il numero e la complessità stessa delle manifestazioni.
Per di più questa sindrome porta in sé uno straordinario paradosso: il caso vuole che l'unico modo per essere allertati sulla sua presenza sia l'avere un arresto cardiaco. Ma andiamo per ordine.
Sindrome di Brugada: Che cos'è
La Sindrome di Brugada fu descritta per la prima volta nel 1988 da autori italiani sul “Giornale Italiano di Cardiologia” e prende il nome dai fratelli Brugada che nel 1992 la introdussero come un'entità clinica distinta.
Solo nel 2002 sono stati definiti i criteri diagnostici e nel 2005 l'attenzione è stata spostata sulla stratificazione del rischio e sulle terapie.
È una malattia aritmogena a trasmissione autosomica dominante caratterizzata da un tipico ECG (segno di Brugada con sopraslivellamento del tratto ST in V1 e V3 e blocco di branca destra) e da sincope e/o morte improvvisa.
In parole semplici è un'aritmia causata da una mutazione genetica (SCN5A, ma non esclusivamente) presente sul cromosoma 3.
Come si manifesta la Sindrome di Brugada
Questa anomalia è responsabile dell'alterazione dei canali del sodio che regolano proprio il passaggio de sodio e del potassio nelle cellule cardiache, indispensabili per il corretto funzionamento elettrico del cuore (ricordiamo infatti che il muscolo cardiaco funziona in una “doppia modalità“: elettrica e meccanica, dipendenti una dall'altra).
La Sindrome sembra essere responsabile di episodi di FV (fibrillazione ventricolare) idiopatica in una percentuale variabile tra il 3 e il 60% dei casi. Si manifesta più frequentemente in giovani maschi (con rapporto maschi/femmine di 8:1) di età compresa tra 30 e 40 anni, ma è difficile stabilire l'esatta prevalenza nella popolazione ed è altrettanto difficile intervenire prontamente, poiché prettamente asintomatica e in assenza di alterazioni strutturali del cuore.
Episodi di sincope e familiarità per morte cardiaca improvvisa in età giovanile sono elementi indispensabili per una eventuale diagnosi e una fondamentale prevenzione primaria. Infatti la sindrome di Brugada, come già detto, si trasmette come carattere autosomico dominante, ciò significa che una persona affetta presenta un rischio pari al 50% di trasmettere la patologia ai propri figli, indipendentemente dal loro sesso.
La diagnosi di Brugada
La diagnosi si basa su un'attenta valutazione (medica ovviamente) dell'elettrocardiogramma, eseguito a riposo, in corso di attività fisica e durante il riposo notturno.
Un Holter cardiaco potrebbe risultare utile. In alcuni casi l'ECG appare normale e la sindrome risulta evidente solo dopo la somministrazione di determinati farmaci (ajmalina, flecainide, procainamide) che bloccano i canali del sodio.
In altri casi, in soggetti ad esempio colpiti da morte cardiaca improvvisa fortunatamente sventata, è possibile eseguire una diagnosi ancora più accurata mediante la ricerca di mutazioni nel gene SCN5A.
Trattamento terapeutico della Sindrome di Brugada
L'identificazione della mutazione permette di estendere l'analisi del DNA ad altre persone della famiglia, con lo scopo di rilevare soggetti asintomatici, ma portatori della Sindrome. Non esiste al momento una terapia farmacologica sicuramente efficace per la prevenzione degli episodi aritmici (è ancora oggi in discussione l'efficacia di sostanze come la chinidina).
Alcuni gruppi di studi sostengono l'efficacia di un'ablazione trans-catetere con radiofrequenza. Nei pazienti a più alto rischio con un'anamnesi comprendente ECG positivo, familiarità ed episodi sincopali è possibile impiantare un defibrillatore automatico (ICD), un apparecchio simile ad un pacemaker in grado di riconoscere e correggere eventuali episodi di fibrillazione ventricolare o aritmie potenzialmente fatali.
Risulta dunque difficile l'approccio a questa strana sindrome, ma l’informazione potrebbe essere fondamentale. Il solo sapere dell’esistenza di una così bizzarra patologia può essere di aiuto per una fondamentale prevenzione primaria, mera risorsa che potrebbe salvare la vita.
















Commento (0)
Devi fare il login per lasciare un commento. Non sei iscritto ?